Титриметрические методы анализа аналитическая химия. Методы титриметрического анализа. Типы титрования. Аналитическая химия. Требования к реакции титрования
Классификация методов титриметрического анализа
Аналитическая химия
Методы титриметрического анализа можно классифицировать по характеру химической реакции, лежащей в основе определения веществ, и по способу титрования.
По своему характеру реакции, используемые в титриметрическом анализе, относятся к различным типам - реакциям соединения ионов и реакциям окисления - восстановления. В соответствии с этим титриметрические определения можно подразделять на следующие основные методы: метод кислотно-основного титрования (нейтрализации), методы осаждения и комплексообразования, метод окисления - восстановления.
Метод кислотно-основного титрования (нейтрализации). Сюда относятся определения, основанные на взаимодействии кислот и оснований, т.е. на реакции нейтрализации:
Методом кислотно-основного титрования (нейтрализации) определяют количество кислот (алкалиметрия) или оснований (ациди-метрия) в данном растворе, количество солей слабых кислот и слабых оснований, а также веществ, которые реагируют с этими солями. Применение неводных растворителей (спирты, ацетон и т. п.) позволило расширить круг веществ, которые можно определять данным методом.
Методы осаждения и комплексообразования. Сюда относятся титриметрические определения, основанные на осаждении того или иного иона в виде малорастворимого соединения или связывания его в малодиссоциированный комплекс.
Методы окисления - восстановления (редоксиметрия). Эти методы основаны на реакциях окисления и восстановления. Их называют обычно по применяемому титрованному раствору реагента, например:
перманганатометрия, в которой используются реакции окисления перманганатом калия KMnO4;
иодометрия, в которой используются реакции окисления иодом или восстановления I-ионами;
бихроматометрия, в которой используются реакции окисления бихроматом калия К2Сr2О7;
броматометрия, в которой используются реакции окисления броматом калия КВrO3.
К методам окисления - восстановления относятся также цериметрия (окисление Се4+-ионами), ванадатометрия (окисление VO3-ионами), титанометрия (восстановление Т13+-ионами). По способу титрования различают следующие методы.
Метод прямого титрования. В этом случае определяемый ион титруют раствором реагента (или наоборот).
Метод замещения. Этот метод применяют тогда, когда по тем или иным причинам трудно определить точку эквивалентности, например при работе с неустойчивыми веществами и т. п.
Метод обратного титрования (титрование по остатку). Этот метод применяют, когда нет подходящего индикатора или когда основная реакция протекает не очень быстро. Например, для определения CaCO3 навеску вещества обрабатывают избытком титрованного раствора соляной кислоты:
Каким бы из методов ни проводилось определение, всегда предполагается:
1) точное измерение объемов одного или обоих реагирующих растворов;
2) наличие титрованного раствора, при помощи которого проводят титрование;
3) вычисление результатов анализа.
В соответствии с этим, прежде чем переходить к рассмотрению отдельных методов титриметрического анализа, остановимся на измерении объемов, расчете концентраций и приготовлении титрованных растворов, а также на вычислениях при титриметрических определениях.
Точка эквивалентности
Точка эквивалентности (в титриметрическом анализе) - момент титрования, когда число эквивалентов добавляемого титранта эквивалентно или равно числу эквивалентов определяемого вещества в образце. В некоторых случаях наблюдают несколько точек эквивалентности, следующих одна за другой, например, при титровании многоосновных кислот или же при титровании раствора, в котором присутствует несколько определяемых ионов.
На графике кривой титрования присутствует одна или несколько точек перегиба, соответствующих точкам эквивалентности.
Точкой окончания титрования (подобна точке эквивалентности, но не то же самое) считают момент, при котором индикатор изменяет свой цвет при колориметрическом титровании.
Методы определения точки эквивалентности
С помощью индикаторов
Это вещества, изменяющие свой цвет вследствие протекания химических процессов. Кислотно-основные индикаторы, например фенолфталеин, изменяют свой цвет в зависимости от pH раствора, в котором они находятся. Редокс-индикаторы изменяют свой цвет вслед за изменением потенциала системы, используются таким образом при окислительно-восстановительном титровании. Перед началом титрования в исследуемый раствор добавляют несколько капель индикатора и начинают по каплям добавлять титрант. Как только раствор вслед за индикатором изменяет свой цвет, титрование прекращают, этот момент приблизительно и есть точка эквивалентности.
Правило выбора индикатора - при титровании используется такой индикатор, который изменяет свою окраску около точки эквивалентности, т.е. интервал перехода окраски индикатора должен по возможности совпадать со скачком титрования.
Потенциометрия
В данном случае используют прибор для измерения электродного потенциала раствора. При достижении точки эквивалентности потенциал рабочего электрода резко изменяется.
С помощью pH-метров
pH-метр по сути своей также является потенциметром, в котором используется электрод, потенциал которого зависит от содержания в растворе ионов H+, это пример использования ионоселективного электрода. Таким образом можно следить за изменением pH в течение всего процесса титрования. При достижении точки эквивалентности pH резко изменяется. Данный способ более точный по сравнению с титрованием с использованием кислотно-основных индикаторов, и может быть легко автоматизирован.
Проводимость
Проводимость раствора электролитов зависит от находящихся в нем ионов. Во время титрования проводимость часто значительно изменяется (Например, при кислотно-основном титровании, ионы H+ и OH− взаимодействуют, образуя нейтральную молекулу H2O, что вызывает изменение проводимости раствора). Общая проводимость раствора зависит и от других присутствующих ионов (например, противоинов), которые вносят в нее различный вклад. Он, в свою очередь, зависит от подвижности каждого иона и от общей концентрации ионов (ионной силы). В связи с этим предсказать изменение проводимости гораздо сложнее, нежели измерить ее.
Изменение цвета
При протекании некоторых реакций происходит изменение цвета и без добавления индикатора. Чаще всего это наблюдается при окислительно-восстановительном титровании, когда исходные вещества и продукты реакции имеют разные цвета в разных степенях окисления.
Осаждение
Если во время реакции образуется твердое нерастворимое вещество, то по окончании титрования образуется преципитат. Классическим примером такой реакции является образование крайне нерастворимого хлористого серебра AgCl из ионов Ag+ и Cl−. Удивительно, но это не позволяет точно определить момент окончания титрования, поэтому осадительное титрование чаще всего используют в качестве обратного титрования.
Изотермическое калориметрическое титрование
Используется изотермический титровальный калориметр, который по величине тепла, которое выделила или поглотила реагирующая система, определяет точку эквивалентности. Данный способ важен в биохимическом титровании, например, для определения того, как ферментный субстрат связывается с ферментом.
Термометрическая титриметрия
Термометрическая титриметрия - чрезвычайно гибкая техника. Она отличается от калориметрической титриметрии тем, что теплота реакции, о которой свидетельствует падение или рост температуры, не используется для определения количества содержащегося в исследуемом образце раствора вещества. Напротив, точка эквивалентности определяется на основе области, в которой происходит изменение температуры. В зависимости от того, является реакция между титрантом и исследуемым веществом экзотермической или эндотермической, температура в течение процесса титрования будет, соответственно, возрастать или падать. Когда все исследуемое вещество прореагировало с титрантом, изменение области, в которой происходит рост или падение температуры, позволяет определить точку эквивалентности и изгиб на кривой температуры. Точно точку эквивалентности можно определить, взяв вторую производную кривой температуры: четкий пик будет указывать на точку эквивалентности.
Спектроскопия
Точку эквивалентности можно определить, измеряя абсорбцию света раствором во время титровании, если известен спектр продукта, титранта или исследуемого вещества. Относительное содержание продукта реакции и исследуемого вещества позволяют определить точку эквивалентности. При этом присутствие свободного титранта (указывающее на завершение реакции) можно обнаружить при очень малых величинах.
Амперометрия
Метод, позволяющий определить точку эквивалентности по величине тока при заданном потенциале. Величина тока вследствие реакции окисления/восстановления исследуемого вещества или продукта у рабочего электрода зависит от их концентрации в растворе. Точке эквивалентности соответствует изменение величины тока. Данный метод наиболее полезен, когда необходимо уменьшить расход титранта, например, при титровании галидов ионом Ag+.
Прямое и обратное титрование.
В простейшем варианте титрования анализируемое вещество взаимодействует непосредственно с титрантом. Количество анализируемого вещества рассчитывают исходя из молярной концентрации титранта, его объема, требуемого для достижения точки эквивалентности, и стехиометрии реакции между определяемым веществом и титрантом.
В обратном титровании анализируемое вещество взаимодействует не с титрантом, а с другим реагентом, присутствующим в избытке. Избыток затем определяют титрованием. Если известно исходное количество реагента и определен его избыток, то разность между ними – это количество реагента, пошедшее на реакцию с определяемым веществом.
Обратное титрование используют, например, когда константа равновесия реакции прямого титрования слишком мала. Среди других причин применения обратного титрования – отсутствие подходящего метода индикации или недостаточная скорость реакции при прямом титровании.
Заместительное титрование.
К анализируемому раствору, содержащему определяемые ионы металла, добавляют магниевый комплекс MgY2-. Т.к. он менее устойчив, чем комплекс определяемого иона металла с комплексоном, то идет реакция замещения и выделяется ион Mg2+.
Затем ион Mg2+ оттитровывают комплексоном III в присутствии эриохрома черного Т.
По объему ЭДТА, затраченному на титрование, рассчитывают массу определяемого иона металла. Такой способ титрования возможен только в случае, если комплексные соединения определяемых металлов устойчивее магниевого комплекса.
___________________________________________________________________________________________________________________________________________________________________________________________
Цель работы: приобретение навыков в применении одного из методов количественного анализа – титриметрического, и обучение элементарным приемам статистической обработки результатов измерений.
Теоретическая часть
Титриметрический анализ - это метод количественного химического анализа, основанный на измерении объема раствора реактива с точно известной концентрацией, расходуемого для реакции с определяемым веществом.
Титриметрическое определение вещества проводится титрованием - добавлением одного из растворов к другому небольшими порциями и отдельными каплями при постоянном фиксировании (контроле) результата.
Один их двух растворов содержит вещество в неизвестной концентрации и представляет собой анализируемый раствор.
Второй раствор содержит реагент с точно известной концентрацией и называется рабочим раствором, стандартным раствором или титрантом.
Требования к реакциям, применяемым при титриметрическом анализе:
1. Возможность фиксировать точку эквивалентности, наиболее широко используют наблюдение за его окраской, которая может меняться при следующих условиях:
Одно из реагирующих веществ окрашено, и окрашенный реагент в процессе реакции изменяет свой цвет;
Применяемые вещества – индикаторы - изменяют окраску в зависимости от свойств раствора (например, в зависимости от реакции среды).
2. Количественное течение реакции, вплоть до равновесия, характеризуемого соответствующей величиной константы равновесия
3. Достаточная скорость химической реакции, т.к. фиксировать точку эквивалентности при медленно текущих реакциях крайне трудно.
4. Отсутствие побочных реакций, при которых точные вычисления невозможны.
Методы титриметрического анализа можно классифицировать по характеру химической реакции, лежащей в основе определения веществ: кислотно-основного титрования (нейтрализации), осаждения, комплексообразования, окисления-восстановления.
Работа с растворами .
Мерные колбы предназначены для измерения точного объема жидкости. Они представляют собой круглые плоскодонные сосуды с узким длинным горлом, на котором имеется метка, до которой следует наполнять колбу (рис. 1).
Рис.1 Мерные колбы
Техника приготовления растворов в мерных колбах из фиксаналов.
Для приготовления раствора из фиксанала ампулу разбивают над воронкой, вставленной в мерную колбу, содержимое ампулы смывают дистиллированной водой; затем растворяют его в мерной колбе. Раствор, находящийся в мерной колбе, доводят до метки. После доведения уровня жидкости до метки раствор в колбе хорошо перемешивают.
Бюретки представляют собой тонкие стеклянные трубки, градуированные в миллилитрах (рис. 2). К нижнему, слегка суженному концу бюретки припаян стеклянный кран или присоединен резиновый шланг с шариковым затвором и стеклянным носиком. Для работы выбирают бюретку в зависимости от объема раствора, применяемого в анализе.

Рис.2. Бюретки
Порядок работы с бюреткой
1. Бюретку промывают дистиллированной водой.
2. Подготовленную к работе бюретку закрепляют вертикально в штативе, с помощью воронки наливают в бюретку раствор так, чтобы его уровень был выше нулевой отметки.
3. Из нижнего оттянутого конца бюретки удаляют пузырьки воздуха. Для этого отгибают его кверху и выпускают жидкость до тех пор, пока весь воздух не будет удален. Затем опускают капилляр вниз.
4. Уровень жидкости в бюретке устанавливают на нулевое деление.
5. При проведении титрования нажимают на резиновую трубку сбоку от шарика и сливают жидкость из бюретки в колбу, вращая последнюю. Сначала титрант, находящийся в бюретке, сливают тонкой струйкой. Когда же окраска индикатора в месте падения капель титранта начнет изменяться, раствор приливают осторожно, по каплям. Титрование прекращают, когда наступает резкое изменение окраски индикатора от приливания одной капли титранта, и записывают объем израсходованного раствора.
6. По окончании работы титрант из бюретки сливают, бюретку промывают дистиллированной водой.
Метод кислотно-основного титрования (нейтрализации)
Метод кислотно-основного титрования основан на реакции взаимодействия кислот и оснований, т.е. на реакции нейтрализации:
H + + OH¯ = H 2 O
При выполнении данного задания используется метод кислотно-основного титрования, основанный на применении реакции нейтрализации:
2NaOH + H 2 SO 4 = Na 2 SO 4 + 2H 2 O
Метод заключается в том, что к раствору определяемого вещества - гидроксида натрия – постепенно прибавляют раствор серной кислоты известной концентрации. Добавление раствора кислоты продолжают до тех пор, пока его количество не станет эквивалентным количеству реагируемого с ним гидроксида натрия, т.е. до нейтрализации щёлочи. Момент нейтрализации устанавливают по изменению окраски индикатора, прибавляемого в титруемый раствор. По закону эквивалентов в соответствии с уравнением:
С н(к-ты) · V (к-ты) = C н (щелочи) · V (щелочи)
С н(к-ты) и C н (щелочи) – молярные концентрации эквивалентов реагирующих растворов, моль/л;
V (к-ты) и V (щелочи) – объёмы реагирующих растворов, л (мл).

С (NaOH) и  - молярные концентрации эквивалента NaOH и H 2 SO 4 в реагирующих растворах, моль/л;
- молярные концентрации эквивалента NaOH и H 2 SO 4 в реагирующих растворах, моль/л;
V (NaOH) и  ) - объёмы реагирующих растворов щёлочи и кислоты, мл.
) - объёмы реагирующих растворов щёлочи и кислоты, мл.
Примеры решения задач.
1. На нейтрализацию 0,05 л раствора кислоты израсходовано 20 см 3 0,5н раствора щелочи. Чему равна нормальность кислоты?
2. Сколько и какого вещества останется в избытке, если к 60см 3 0,4н раствора серной кислоты прибавить 120см 3 0,3н раствора гидроксида калия?
Решение задач по определению рН раствора, концентраций различного типа представлено в методическом пособии .
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Получите у лаборанта колбу с раствором щёлочи неизвестной концентрации. Пробы анализируемого раствора отмерьте мерным цилиндром по 10 мл в три конические колбы для титрований. В каждую из них добавьте 2-3 капли индикатора метилового оранжевого. Раствор приобретёт жёлтую окраску (метилоранж жёлтый в щелочной среде и оранжево-красный в кислой).
Приготовьте к работе установку для титрований (рис.3) Бюретку ополосните дистиллированной водой, а затем заполните раствором серной кислоты точно известной концентрации (молярная концентрация эквивалента H 2 SO 4 указана на склянке) выше нулевого деления. Каучуковую трубку со стеклянным наконечником отогните вверх и, оттягивая резину от стеклянной оливы, закрывающей выход из бюретки, медленно выпускайте жидкость так, чтобы после заполнения наконечника в нём не осталось пузырьков воздуха. Избыток раствора кислоты выпустите из бюретки в подставленный стакан, при этом нижний мениск жидкости в бюретке должен установиться на нулевом делении.
Одну из колб раствора щёлочи подставьте под наконечник бюретки на лист белой бумаги и приступайте непосредственно к титрованию: одной рукой медленно подавайте кислоту из бюретки, а другой непрерывно перемешивайте раствор круговым движением колбы в горизонтальной плоскости. В конце титрования раствор кислоты из бюретки следует подавать по каплям до тех пор, пока от одной капли раствор примет неисчезающую оранжевую окраску.
Определите объём кислоты, израсходованный на титрование, с точностью до 0,01мл. Отсчёт делений бюретки производите по нижнему мениску, при этом глаз должен находиться на уровне мениска.
Повторите титрование ещё 2 раза, начиная каждый раз с нулевого деления бюретки. Результаты титрований запишите в таблицу 1.
Концентрацию раствора щёлочи вычислите по формуле:

Таблица 1
Результаты титрования раствора гидроксида натрия
Проведите статистическую обработку результатов титрований по методике, описанной в приложении. Результаты статистической обработки экспериментальных данных сведите в таблицу 2.
Таблица 2
Результаты статистической обработки экспериментальных данных титрования раствора гидроксида натрия. Доверительная вероятность α = 0,95.
| n | S x |  |
||
Запишите результат определения молярной концентрации эквивалента NaOH в анализируемом растворе в виде доверительного интервала.
ВОПРОСЫ ДЛЯ САМОКОНТРОЛЯ
1. Раствор гидроксида калия имеет рН =12. Концентрация основания в растворе при 100% диссоциации равна … моль/л.
1) 0,005; 2) 0,01; 3) 0,001; 4) 1·10 -12 ; 5) 0,05.
2. На нейтрализацию 0,05 л раствора кислоты израсходовано 20 см 3 0,5 н раствора щелочи. Чему равна нормальность кислоты?
1) 0,2 н; 2) 0,5 н; 3) 1,0 н; 4) 0,02 н; 5) 1,25 н.
3. Сколько и какого вещества останется в избытке, если к 75 см 3 0,3 н раствора серной кислоты прибавить 125 см 3 0,2 н раствора гидроксида калия?
1) 0,0025 г щелочи; 2) 0,0025 г кислоты; 3) 0,28 г щелочи; 4) 0,14 г щелочи; 5) 0,28 г кислоты.
4. Метод анализа, основанный на определении повышения температуры кипения, называется…
1) спектрофотометрический; 2) потенциометрический; 3) эбулиоскопический; 4) радиометрический; 5) кондуктометрический.
5. Определить процентную концентрацию, молярность и нормальность раствора серной кислоты, полученного при растворении 36 г кислоты в 114 г воды, если плотность раствора 1,031 г/см 3 .
1) 31,6 ; 3,77; 7,54 ; 2) 31,6; 0,00377; 0,00377 ;
3) 24,0 ; 2,87; 2,87 ; 4) 24,0 ; 0,00287; 0,00287;
5) 24,0; 2,87; 5,74.
Титриметрический анализ
История и принцип метода
Титриметрический анализ (титриметрия) -важнейший из химических методов анализа. Он возник в XVIII веке, вначале как эмпирический способ проверки качества различных материалов, например, уксуса, соды, отбеливающих растворов. На рубеже XVIII и XIX веков были изобретены бюретки и пипетки (Ф.Декруазиль). Особое значение имели труды Ж.Гей-Люссака, который ввел основные термины этого метода: титрование, титрант и другие, происходящие от слова «титр». Титр – это массарастворенного вещества (в граммах), содержащаяся в одном миллилитре раствора. Во времена Гей-Люссака результаты анализа вычисляли именно с помощью титров. Однако титр как способ выражения концентрации раствора оказался менее удобным, чем другие характеристики (например, молярные концентрации), поэтому в современной аналитике химии расчеты с применением титров ведут довольно редко. Напротив, различные термины, произведенные от слова «титр», применяют очень широко.
В середине XIX века немецкий химик К.Мор обобщил все созданные к тому времени титриметрические методики и показал, что в основе любой методики лежит один и тот же принцип. К раствору пробы, содержащей определяемый компонент Х, всегда прибавляют раствор с точно известной концентрацией реагента R (титрант). Этот процесс и называют титрованием. Проводя титрование, аналитик следит за протеканием химической реакции между Х и добавляемым R . По достижении точки эквивалентности (т.экв.), когда число молей эквивалентов введенного R точно сравняется с числом молей эквивалентов находившегося в пробе вещества Х, титрование прекращают и измеряютобъем затраченного титранта. Момент окончания титрования называют конечной точкой титрования (к.т.т.), ее, как и т.экв., выражают в единицах объема, обычно в миллилитрах. В идеальном случае V к.т.т = V т.экв. , но на практике точное совпадение по разным причинам не достигается, титрование заканчивают чуть раньше или, наоборот, чуть позже, чем будет достигнута т.экв. Естественно, титрование следует проводить так, чтобы различие между V т.экв. и V к.т.т. было бы как можно меньшим.
Поскольку массу или концентрацию Х рассчитывают по объему титранта, затраченному на титрование пробы (по V к.т.т.), впрошлом титриметрию называли объемным анализом . Это название нередко используют и сегодня, но термин титриметрический анализ более точен. Дело в том, что операция постепенного прибавления реагента (титрование) характерна для любой методики этого типа, а расход титранта можно оценивать не только путем измерения объема, но и другими способами. Иногда добавляемый титрант взвешивают (измерение массы на аналитических весах дает меньшую относительную погрешность, чем измерение объема). Иногда измеряют время, за которое будет введен титрант (при постоянной скорости ввода).
С конца XIX века титриметрические методики стали применять и в исследовательских,и в заводских, и в других лабораториях. С помощью нового метода оказалось возможным определять миллиграммовые и даже микрограммовые количества самых разных веществ. Широкому использованию титриметрии способствовали простота метода, невысокая стоимость и универсальность оборудования. Особенно широко титриметрию стали применять в 50-х годах XX века,после создания швейцарским аналитиком Г.Шварценбахомнового варианта этого метода (комплексонометрии). Одновременно началось широкое применение инструментальных методов контроля к.т.т. К концу 20 века значение титриметрии несколько снизилось в связи с конкуренцией более чувствительных инструментальных методов, но и сегодня титриметрия остается очень важным методом анализа. Она позволяет быстро, легко и достаточноточно определять содержание большинства химических элементов, отдельные органические и неорганические вещества, суммарное содержание однотипных веществ, а также обобщенные показатели состава (жесткость воды, жирность молока, кислотность нефтепродуктов).
Техника проведения титриметрического анализа
Принцип метода станет более понятен после изложения техники его проведения. Итак, пустьВам принесли раствор щелочи неизвестной концентрации, и Ваша задача – установить его точную концентрацию. Для этого Вам понадобится раствор регента , или титранта – вещества, которое вступает в химическую реакцию со щелочью, причем концентрация титранта должна быть точно известна. Очевидно, что для установления концентрации щелочи в качестве титранта используемраствор кислоты.
1. Отбираем с помощью пипетки точный объем анализируемого раствора – он называется аликвота . Как правило, объем аликвоты составляет 10-25 мл.
2. Переносим аликвоту в колбу для титрования, разбавляем водой и добавляем индикатор.
3. Заполняем бюретку раствором титранта и выполняем тирование – медленное, по каплям, добавление титранта к аликвоте исследуемого раствора.
4. Заканчиваем титрование в момент, когда индикатор изменит свою окраску. Этот момент называется конечной точкой титрования – к.т.т. К.т.т., как правило, совпадает с моментом, когда реакция между определяемым веществом и титрантом закончена, т.е. к аликвоте добавлено точно эквивалентное количество титранта – этот момент называется точкой эквивалентности, т.э. Таким образом т.э. и к.т.т. – это две характеристики одного и того же момента, одна – теоретическая, другая – экспериментальная, зависящая от выбранного индикатора. Поэтому надо правильно выбирать индикатор, с тем, чтобы к.т.т. как можно точнее совпадала с т.э.
5. Измеряют объем титранта, пошедшего на титрование, и вычисляют концентрацию исследуемого раствора.
Виды титриметрического анализа
Классифицировать титриметрические методики можно по нескольким независимым признакам: а именно: 1) по типу реакции между Х и R , 2) по способу проведения титрования и расчета результатов,3) по способу контроля т.экв.
Классификация по типу химической реакции – наиболее важная. Напомним, что далеко не все химические реакции можно использовать для проведения титрований.
Во-первых, как и в других химических методах, определяемый компонент (аналит) должен количественно реагировать с титрантом.
Во-вторых, надо, чтобы равновесие реакции устанавливалось как можно быстрее. Реакции, в которых после добавления очередной порции титранта установление равновесия требует хотя бы нескольких минут, в титриметрии применять затруднительно или вообще невозможно.
В-третьих, реакция должна отвечать единственному и заранее известному стехиометрическому уравнению. Если реакция ведет к смеси продуктов, состав этой смеси будет меняться в ходе титрования и зависеть от условий проведения реакции. Зафиксировать точку эквивалентности будет очень трудно, а результат анализа окажетсянеточным.Совокупности указанных требований отвечают реакции протолиза (нейтрализации), многие реакции комплексообразования и окисления-восстановления, а также некоторые реакции осаждения. Соответственно в титриметрическом анализе выделяют:
Метод нейтрализации,
Комплексометрию,
Редоксметрические методы
Методы осаждения.
Внутри каждого метода выделяют отдельные его варианты (табл.1). Их названия происходят от наименований реагентов, используемых в каждом из вариантов в качестве титранта (перманганатометрия, иодометрия, хроматометрия и т.п.).
Таблица 1.
Классификация титриметрических методик по типу используемой химической реакции
|
Реакция |
Метод |
Реагент (титрант) |
Вариантметода |
Определяемыевещества |
|
Протолиз |
Методнейтрализации |
Н Cl, HClO 4 , HNO 3 |
Ацидиметрия |
Oc нования |
|
KOH, NaOH и др. |
Алкалиметрия |
Кислоты |
||
|
Комплексо-образование |
Комплексо-метрия |
ЭДТА |
Комплексонометрия |
Металлы и ихсоединения |
|
Фторидометрия, цианидометрия |
Некоторые металлы, органическиевещества |
|||
|
Окисление-восстанов-ление |
Редокс-метрия |
KMnO 4 К 2 С r 2 O 7 |
Перманганатометрия хроматометрия |
Восстановители |
|
KJ и Na 2 S 2 O 3 |
Иодометрия |
Восстановители,окислители, кислоты |
||
|
Аскорбиновая кислота |
Аскорбинометрия |
Окислители |
||
|
Осаждение |
Седиметрия |
AgNO 3 |
Аргентометрия |
Галогениды |
|
Hg 2 (NO 3) 2 |
Меркуриметрия |
|||
|
KSCN |
Роданометрия |
Некоторые металлы |
||
|
Ba(NO 3) 2 |
Бариеметрия |
Сульфаты |
Классификация по способу титрования. Обычно выделяют три способа: прямое, обратное и заместительное титрование. Прямое титрование предполагает непосредственное прибавление титранта к раствору пробы. Иногда применяют другой порядок смешивания реагентов – к известному количеству R постепенно добавляют раствор пробы, в котором хотят определить концентрацию Х; но это тоже прямое титрование. В обоих случаяхрасчет результатов анализа ведут по одним и тем же формулам, основанным на законе эквивалентов.
ν Х = ν R
где ν Х иν R – количества молей эквивалентов Х и R . Расчетные формулы, основанные на соотношении, а также примеры расчетов будут даны ниже.
Прямое титрование - удобный и самый распространенный вариант титриметрии. Он более точен, чем другие. Ведь случайные погрешности в основном возникают при измерении объема растворов, а в данном способе титрования объем измеряют только один раз.Однако прямое титрование возможно далеко не всегда. Многие реакции между Х и R идут недостаточно быстро, и после добавления очередной порции титранта в растворе не успевает установиться равновесие. Иногда прямое титрование невозможно из-за побочных реакций или ввиду отсутствия подходящего индикатора. В подобных случаях применяют более сложные схемы обратного или заместительного титрования. Они включают не менее двух химических реакций.
Обратное титрование проводят по двухстадийной схеме:
Х + R 1 =Y 1
R 1 + R 2 = Y 2
Вспомогательный реагент R 1 вводят в точно известном количестве. Объем и концентрацию раствора R 1 выбирают так, чтобы R 1 после завершения реакции с Хостался в избытке. Затем непрореагировавшую часть R 1 оттитровывают титрантом R 2 . Примером может быть перманганатометрическое титрование органических веществ. Титровать многие веществаперманганатом «напрямую» не удается из-за замедленности их окисления и по другим причинам. Но можно сначала добавить к анализируемой пробе известное (избыточное) количество KMnO 4 , подкислить и нагреть полученный раствор. Это приведет к полному и быстрому завершению окисления органических веществ. Затем оттитровывают оставшийся перманганат каким-либо активным восстановителем, например, раствором SnCl 2 или FeSO 4 .
Расчет результатов обратного титрования проводят, исходяиз очевидного соотношения:
ν Х =ν R 1 - ν R 2
Поскольку объемы в данном случае измеряют два раза (сначала объем раствора реагента R 1 , затем объем титранта R 2), случайная погрешность результата анализа несколько выше, чем при прямом титровании. Особенно сильно возрастает относительная погрешность анализа при малом избытке вспомогательного реагента, когдаν R 1 ≈ν R 2 .
Классификацияпо способу контроля т.экв. Известно несколько таких способов. C амый простой - безындикаторное титрование, самый распространенный – титрование с цветными индикаторами, а самые точные и чувствительные –инструментальные варианты титриметрии.
Безындикаторное титрование основано на применении реакций, которые сопровождаются изменением видимых свойств титруемого раствора. Как правило, один из реагентов (Х или R ) имеет видимую окраску. Ход такой реакции контролируют без специальных приборов и без добавления реактивов-индикаторов. Так, бесцветные восстановители титруют в кислой среде фиолетовым раствором окислителя – перманганата калия (KMnO 4). Каждая порция добавляемого титрантабудет сразу же обесцвечиваться, превращаясь под действием восстановителя в ионы Mn 2+ . Так будет продолжаться вплоть до т.экв. Однако первая же «лишняя» капля титранта окрасит титруемый раствор врозово-фиолетовый цвет, окраска не исчезнет и при перемешивании раствора. При появлении неисчезающей окраски титрование прекращают иизмеряют объемзатраченного титранта (V к.т.т.). Конец титрованияможно зафиксировать не только по появлению окраски титруемого раствора, как в рассмотренном примере, но ипо обесцвечиванию ранее окрашенного раствора пробы, а также по появлению какого-либо осадка, его исчезновению или изменению внешнего вида. Безындикаторное титрование применяют довольно редко, так как лишь немногие реакции сопровождаются изменением видимых свойств раствора.
Инструментальное титрование . За протеканием реакции между Х и R можно следить не просто «на глаз» (визуально), но и с помощью приборов, измеряющих некоторое физическое свойство раствора. Варианты инструментальной титриметрии различают, смотря по тому, какое именно свойство раствора контролируется. Можно использовать любое свойство, зависящее от качественного и количественного состава титруемого раствора. А именно, можно измерять электропроводность раствора (этот вариант называют кондуктометрическим титрованием), потенциал индикаторного электрода, опущенного в титруемый раствор (потенциометрическое титрование), поглощение света титруемым раствором (фотометрическое титрование) и т.п.Прекратить титрование можно тогда, когдабудет достигнуто некоторое заранее выбранное значение измеряемого свойства. Например, титруют раствор кислоты щелочью до тех пор, пока не будет достигнуто значение рН = 7. Однако чаще поступают по-другому - выбранное свойство растворамногократно (или даже непрерывно) измеряют по мере ввода титранта, причем не только до, но и после ожидаемой т.экв.По полученным данным строят графическую зависимость измеренного свойства от объема добавленного титранта (кривую титрования ). Вблизи точки эквивалентностинаблюдается резкое изменение составаисвойств титруемого раствора, а на кривой титрования регистрируется скачок или излом. Например, скачок потенциала электрода, опущенного в раствор. Положение т.экв оценивают по положению перегиба на кривой. Такой вариант анализа более трудоемок и длителен, чем обычное титрование, но дает более точные результаты. За одно титрование удается определить по отдельности концентрации целого ряда компонентов.
Известно более десятка вариантов инструментальной титриметрии. В создании их важную роль сыграл американский аналитик И.Кольтгоф. Соответствующие методики различаются по измеряемому свойству раствора, по используемой аппаратуре и по аналитическим возможностям, но все они чувствительнее и селективнее, чеминдикаторные илибезындикаторные визуальные варианты титриметрии. Инструментальный контроль особенно важен, когда нельзя применять индикаторы, например, при анализе мутных или интенсивно окрашенных растворов, а также при определении микропримесей и при анализе смесей. Однако инструментальная титриметрия требует оснащения лаборатории специальными приборами, желательно - самопишущими или полностью автоматизированными, что не всегда экономически целесообразно. Во многих случаях достаточно точные и надежные результаты могут быть получены более простым и дешевым способом, основанным на применении индикаторов.
Использование индикаторов . К титруемой пробе можно заранее добавить небольшое количество специального реактива - индикатора . Титрование надо будет прекратить в тот момент, когда индикатор под действием введенного титранта изменит видимую окраску, это и есть конечная точка титрования. Важно, чтобы изменение окраски происходило не постепенно, ав результате добавления всего одной «лишней» капли титранта. В некоторых случаях индикатор меняет не свою окраску, арастворимость или характер свечения. Однако такие индикаторы (адсорбционные, флуоресцентные, хемилюминесцентные и др.) применяют намного реже, чемцветные индикаторы. Изменение окраски любого индикатора происходит благодаря химическому взаимодействию индикатора с титрантом, приводящему кпереходу индикаторав новую форму.Свойства индикаторов необходимо рассмотреть более детально.
Индикаторы
В аналитических лабораториях применяют несколько сот цветных индикаторов разного типа (кислотно-основные, металлохромные, адсорбционные и т.п.). Когда-то в качестве индикаторов использовались настойки, полученные из растений - из цветов фиалки или из особого вида лишайников (лакмус). Впервые такие индикаторы стал применять еще Р.Бойль. В настоящее время природные индикаторы не используют, поскольку они всегда являются смесью разных веществ, поэтому переход их окраски выражен недостаточно четко. Современные индикаторы – это специально синтезированные индивидуальные органические соединения. Как правило, индикаторами являются соединения ароматического ряда, молекулы которых содержат несколько функциональных групп (заместителей).Известно множество подобных соединений, но только некоторые из нихможно применять в качестве цветных индикаторов. Предполагаемый индикатор должен отвечать целому ряду требований:
· индикатор должен хорошо растворяться, даваярастворы, устойчивые при хранении;
· в растворе индикатор должен существовать в нескольких формах, различных по структуре молекулы. Между формами должно устанавливаться подвижноехимическое равновесие. Например, кислотная форма индикатора переходит в основную (и обратно),окисленная- ввосстановленную (и обратно); металлохромный индикатор обратимо связываетсяв комплекс с ионами металла, и т.п.;
· цветной индикатордолжен интенсивно поглощать свет в видимой области спектра. Окраска его раствора должна быть различима даже при очень низкой концентрации (10 -6 – 10 -7 моль/л). В этом случае можно будет вводить в титруемый раствор очень малые количества индикатора, что способствует получению более точных результатов анализа;
· разныеформыиндикаторадолжны быть различны по своей окраске, то есть по спектру поглощения в видимой области. В таком случае в ходе титрования будет наблюдаться контрастный цветовой переход.Например, переход окраски индикатора из розовой в изумрудно-зеленую хорошо заметенна глаз. Зафиксировать же конечную точку титрования (к.т.т.) по переходурозовой окраски воранжевую или фиолетовую гораздо труднее. Очень важно, насколько различны спектры поглощения двух форм индикатора. Если одна из форм индикатора максимально поглощает свет с длиной волны λ 1 , а другая- с длиной волны λ 2 , то разность∆λ = λ 1 - λ 2 характеризует контрастность цветового переход. Чем больше ∆λ, тем лучше воспринимается на глаз переход окраски индикатора. Для повышения визуальной контрастности цветового перехода иногда используют смеси разных индикаторов или к индикатору добавляют посторонний инертный краситель;
· переход индикатора из одной формы в другую при изменении состава раствора должен проходить очень быстро, за доли секунды;
· переход должен вызываться единственным фактором, одним и тем же у всех индикаторов данного типа. Так, изменение окраски кислотно-основного индикатора не должно происходить за счет реакций другого типа, например при взаимодействии с окислителями, или ионами металлов, или белками! Напротив, редокс-индикаторы должны менять свою окраскутолько вследствие взаимодействия с окислителями и восстановителями, и происходить это должно при определенном потенциале, специфическом для каждого редокс-индикатора. Окраска этих индикаторов и потенциал перехода не должны зависеть от рН раствора. К сожалению, на практике потенциал перехода многих редокс-индикаторов зависит иот рН.
Чтобы ослабить влияние побочных процессов, иногда индикатор не вводят в титруемый раствор, а, наоборот, в ходе титрования периодически отбирают каплю титруемого раствора, смешивают ее на часовом стекле с каплей раствора индикатора и наблюдают, какая окраска получается. Такой прием позволяет использовать необратимо реагирующие индикаторы. С «внешним индикатором» удобнее работать, если заранее пропитать имбумагу.
Конечная точка титрования,фиксируемая по переходу окраски индикатора, может не совпадать с точкой эквивалентности. Несовпадение V к.т.т. и V т.экв приводит к систематической погрешностирезультата анализа. Величина погрешности определяется природой данного индикатора, его концентрацией и составом титруемого раствора.
Принцип подбора индикаторов очень прост и универсален:характеристика перехода индикатора (рТ-показатель титрования, потенциал перехода и т.п.) должна соответствовать ожидаемому составу титруемого растворав точке эквивалентности. Так, если аналитик титрует водный раствор сильной кислоты сильным основанием, в точке эквивалентности раствор будет иметь рН = 7. Следовательно,надо использоватькислотно-основной индикатор, который меняет свою окраску приблизительно при рН 7 (бромтимоловый синий и т.п.).Необходимые сведения о рТ - показателях титрования для индикаторов разного типа есть в справочной литературе.
Расчет результатов титриметрического анализа
Результаты титриметрического анализа не рекомендуется рассчитывать непосредственно по уравнению реакции, например, с помощью пропорций. Такой «школьный» способ решения расчетных задач нерационален и, как правило,не дает требуемой точности. Результаты титриметрического анализа рассчитывают по одной из несколькихготовых алгебраических формул, выведенных на основании закона эквивалентов. Исходными данными будут oбъем затраченного титранта (в миллилитрах) и концентрация титранта (в моль/литр), их надо установить с необходимой точностью.
Способ расчета не зависит от типа химическойреакции, протекающейв ходе титрования, и способа контроляточки эквивалентности (индикатор, прибор и т.п.). Выбор расчетной формулы определяется тем,какойспособ титрования(прямое, обратное, заместительное) применяютв ходе анализа.Выбираяформулу, следует различать два случая:а) расчетконцентрациираствора Х;б) определениемассовой доли компонента (процентного содержанияХ в пробе).
Наиболее просто выглядят расчетные формулы, если концентрацииопределяемогокомпонента и титранта выражают числоммолей их эквивалентов в литре соответствующих растворов, т.е. используют концентрации определяемого компонента (N x ) и титранта (N T ), выраженные числом молей эквивалента в литре раствора. Ранее эти концентрации называли нормальными. Теперь этот термин применять не рекомендуется, но на практике его используют весьма широко, особенно в редоксметрии. А вот в комплексонометрии и в некоторых других методах, где 1 моль определяемого вещества Х всегда реагирует с 1 молем титранта, нормальные концентрации совпадают с обычными молярными концентрациями (C x и С Т ), а поэтому при расчете результатов нормальные концентрации и эквиваленты применять незачем.
В отличие от обычных молярных концентраций, нормальная концентрация определяется с учетом химизма реакции, протекающей в ходе титрования. Полезно запомнить, чтонормальная концентрация Х в растворе либо равнаего молярной концентрации,либо превосходит ее в несколько (2,3,4....)раз,смотря по тому, сколько протонов (или электронов) участвует в реакции, в расчете на одну частицу Х. При записи уравнения реакции, определенииэквивалентов и расчете нормальных концентраций следуетучитывать условия, в которых протекает титрование, и даже выбор индикатора.
Масса оттитрованного Xпри прямомтитровании равна (в мг):
m x =N T . V T . Э x , (1),
где Э x - молярная масса эквивалента Х, соответствующая одному протону (в кислотно-основных реакциях),одному электрону (в окислительно-восстановительных реакциях),одномулиганду (в реакциях комплексообразования), и т.п. V T – объем титранта (в мл). В комплексонометриимассу определяемого вещества (в мг) лучше рассчитывать по формуле, в которую входит величина М х -молярная масса Х:
m x = С T . V T . М x (2).
Из (4.11) следует, что массовая доля Х в навеске пробы, выраженная в %, равна:
%X = N T . V T . Э x . 100 % / m S , (3),
где m S - масса навески в мг.Обычно результат титрования не зависит от того, в какомобъеме воды растворили навеску пробы перед титрованием, и этот объем в расчетах не учитывают. Если же титруют невсю навеску, анекоторую ее часть (аликвоту), то надо учесть дополнительный коэффициент К , равныйотношению V 0 -объема раствора,в который перевелиэту навеску и из которого отбирали аликвоты,к V aliq - объемуодной аликвоты:
m x = К. N T . V T . Э x , (4).
При расчете концентрации по способу прямого (или заместительного) титрованияприменяютпростую формулу, непосредственно следующую из закона эквивалентов:
N х . V х =N T . V T (5).
анализа, однако в заводских лабораториях пользуются и другими способами расчета.
Приготовление рабочих растворов в титриметрии
Применяемые в титриметрическом анализе рабочие растворы точно известной концентрации готовят несколькими способами:
· по точной навеске химического реактива , взятой на аналитических весах. Эту навеску растворяют в небольшом количестве растворителя, а затем в мерной колбе доводят объем полученного раствора до метки. Полученныерастворыназывают стандартными, а соответствующие реактивы – первичнымистандартами. Лишь немногие вещества могут быть первичными стандартами – они должны быть чистыми химическими веществами постоянного и точно известного состава, твердыми при комнатной температуре, устойчивыми на воздухе, не гигроскопичными и не летучими. Примерами могут бытьдихромат калия, комплексон III , щавелевая кислота. Напротив, по навеске нельзя приготовить стандартный раствор соляной кислоты (реактив «соляная кислота» - жидкость с неточно известным составом), хлорида двухвалентного железа (быстро окисляется на воздухе), едкого натра (гигроскопичен) и многих других веществ.
· из фиксаналов . Этим термином называют запаянную стеклянную ампулу, в которой содержится определенное количество реагента, обычно 0,1000 моль эквивалента. Фиксаналы готовят в заводских условиях. Если в лаборатории количественно перенести содержимое фиксанала в мерную колбу на 1000 мл и довести растворителем до метки, получится литр точно 0,1000 н раствора. Приготовление фиксанальных растворов не только экономит время аналитика, но ипозволяет готовить растворы с точно известной концентрацией из таких веществ, которые не обладаюткомплексом свойств, необходимых для первичных стандартов (например, фиксанальные растворы соляной кислоты,аммиака или иода).
· по приблизительно известной навеске химического реактива, взятой на технических весах. Эту навеску растворяют в приблизительно известном количестве растворителя. Затемпроводят дополнительную операцию – стандартизацию полученного раствора. Например, титруют полученным раствором точную навеску другого вещества (первичного стандарта). Можно поступить и по-другому: взять известный объем (аликвоту) приготовленного раствора и оттитровать его подходящим стандартным раствором.По объему, пошедшему на титрование, рассчитывают точную концентрацию приготовленного раствора. Такие растворы называют стандартизованными. Например, раствор КОН стандартизуют по навеске щавелевой кислоты или с помощью фиксанального раствора соляной кислоты. Если вещество в лабораторииимеется в виде концентрированного раствора приблизительно известной концентрации (например, соляная кислота), то вместо его взвешивания отмеривают некоторый, заранее рассчитанный объем концентрированного раствора. Это требует знания плотности исходного раствора. Затем, как и в предыдущем случае, стандартизуют полученный раствор.
Концентрация растворов не должна самопроизвольно изменяться при хранении. В этом случае заранее приготовленные (стандартные или стандартизованные) растворы можно будет использовать для проведения титрованийбез каких-либо дополнительных операций.Следует учесть, что чем более разбавлен раствор, тем, как правило, он менее устойчив при хранении (гидролиз растворенного вещества, его окисление кислородом воздуха, адсорбция на внутренней поверхности стеклянной посуды и др.). Поэтомурабочие растворыс низкой концентрацией, как правило, не готовят заранее. Их готовят лишь по мере надобности, в день употребления. Для этого разбавляют исходные (стандартные, фиксанальные или стандартизованные) растворы чистым растворителем в точно известное число раз (обычно за одну операцию раствор разбавляют в 5 или 10 раз). Если требуются еще более разбавленные растворы, то эту операцию повторяют. Например, из 0,1 М раствора готовят 0,01 М, из того - 0,001 М и т.д.
Приготовление растворов с точно известной концентрацией требует использованияцелого набора специальной мерной посуды, позволяющейизмерять объемы с требуемой точностью. Это мерные колбы, пипетки и бюретки. В руководствахклабораторным работам приводятся описания мерной посудыи правила работы с ней.
Методы титрования
Методотдельных навесок и метод аликвот . Дляуменьшениявлияния случайных погрешностей титрование обычно повторяют несколько раз, а затем усредняют результаты. Повторные анализыможно проводить двумя разными способами:по методу отдельных навесоки по методу аликвот. Оба способа используют и при стандартизации рабочих растворов, и непосредственнов анализереальных объектов.
Метод отдельных навесок , как ясно из его названия, предполагает, что для титрования берут несколько навесок анализируемого материала. Массы их должны быть приблизительно равны. Размер навески выбирают с учетом желаемого расхода титранта на одно титрования (не более объема бюретки) и с учетом концентрации титранта.
Пусть взяты три навески щавелевой кислоты,массы которыхуказаны в табл.2. По данным каждого титрования вычисляют (по отдельности!) концентрацию КОН. Затем усредняютконцентрации.Объемы, затраченные на титрование разных навесок,усреднять нельзя!
Таблица 2.Пример расчета результатов анализа по методу отдельных навесок
|
Номернавески |
Массанавески,мг |
Объемтитранта,мл |
Найденная концентрацияКОН, моль/л |
|
95,7 |
14,9 |
0,102 |
|
|
106,9 |
16,2 |
0,105 |
|
|
80,8 |
12,7 |
0,101 |
|
|
Средний результат анализаС КОН =0,103 моль/л |
|||
Метод титровпанияаликвот (или метод пипетирования) основан на титровании нсекольких отдельных аликвот – небольших объемов исследуемого раствора, отобранных с помощью пипеток.
Метод отдельных навесок и методтитрованияаликвотиспользуют не только при прямом титровании, как это показано в приведенных примерах, но и при обратном, и при заместительном титровании. Выбирая способ титрования, следует учесть, что метод отдельных навесок дает более точные результаты, но он более трудоемкий и требуетбольшего объема расчетов. Поэтому метод отдельных навесок лучше использоватьдля стандартизации рабочих растворов, а для серийно выполняемыханализов применять более экспрессный метод аликвот.
Форма кривых титрования
Логарифмические кривые титрования представляют графическую зависимостьлогарифмаравновеснойконцентрации одного из реагентов от объема добавленного титранта. Вместо логарифма концентрации на вертикальной оси обычно откладывают величину рН раствора (водородный показатель). Применяют и другие аналогичные показатели (например, pAg = - lg ), а также величину тех физико-химических свойств титруемого раствора, которые линейно зависят от логарифмов равновесных концентраций. Примером может быть электродный потенциал (E ).
Если в растворе содержитсятолько одно вещество, реагирующее с титрантом, причемреакция описывается единственным химическим уравнением (то есть проходит не ступенчато)- на логарифмической кривой наблюдается почти вертикальный участок, называемой скачком титрования . Напротив, участкикривой вдали от т.экв. близки к горизонтальным. Примером могут быть зависимости рН растворов от объема V добавленного титранта, показанные на рис.1
Рис.1. Вид кривых титрования
Чем выше высота скачка на кривой тирования, тем точнее можно зафиксировать точку эквивалентности.
Кислотно-основное титрование (метод нейтрализации)
Принцип метода
Метод нейтрализации основан на проведении кислотно-основных (протолитических) реакций. В ходе такого титрования меняетсязначение рН раствора. Кислотно-основные реакции подходят для титриметрического анализа в наибольшей степени: они протекают по строго определенным уравнениям, без побочных процессов и с очень высокой скоростью. Взаимодействие сильных кислот с сильными основаниями приводит к высоким константам равновесия. Для обнаружения к.т.т. существует удобный и хорошо изученный способ - применение кислотно-основных индикаторов. Можно использовать и инструментальные методы, они особенно важны при титровании неводных, мутныхили окрашенных растворов.
Метод нейтрализации включаетдва варианта – ацидиметрию (титрант – раствор сильной кислоты) и алкалиметрию (титрант – раствор сильного основания). Эти методы соответственно применяют для определения оснований и кислот, в том числе ионных и многопротонных. Возможность титрования сильных протолитов определяется их концентрацией; титрование возможно, если С х > 10 - 4 М .В ходе такого титрованияв водном растворе идет реакция:
H 3 O + +OH - ® 2 Н 2 О
Титрование слабых кислот и слабых оснований в водных растворах соответствует схемам:
НА+ОН - ® Н 2 О(алкалиметрия)
В+Н 3 O + ® НВ + + Н 2 О(ацидиметрия)
Примеры практического применения кислотно-основного титрования:
· определение кислотности пищевых продуктов, почв и природных вод (алкалиметрическое титрованиеводных растворов с индикатором фенолфталеином);
· определение кислотности нефтепродуктов (алкалиметрическое титрование неводных растворов с инструментальным контролем к.т.т.);
· определение карбонатов и гидрокарбонатов в минералах и строительных материалах (ацидиметрическое титрование водных растворов с двумя индикаторами);
· определение азота в солях аммония и в органических веществах (метод Кьельдаля). В этом случае органические азотсодержащие вещества разлагают кипячением с концентрированной серной кислотой в присутствии солей ртути, аммонийный азот отгоняют действием щелочи при нагревании, аммиак поглощают стандартным раствором НСl , взятым в избытке. Затем титруют щелочью непрореагировавшую часть НСl в присутствии индикатора метилового оранжевого. В данной методике используют и принцип замещения, и способ обратного титрования.
Рабочие растворы. При ацидиметрическом титровании водных растворовв качестве титрантов используют растворы сильных кислот (НСl , реже НNO 3 или H 2 SO 4). В алкалиметрии титранты - растворы NaOH или КОН. Однако перечисленные реагенты не обладают свойствами, которые позволяли бы готовитьиз них стандартные растворыпросто по точной навеске. Так, твердые щелочи гигроскопичны и всегда содержат примеси карбонатов. В случае НСl и других сильных кислот исходный реактив представляет собой не чистое вещество, а раствор с неточно известной концентрацией. Поэтому в методе нейтрализации вначале готовят раствор с приблизительно известной концентрацией, а потомстандартизуют его. Растворы кислот стандартизуют по безводному карбонату натрия Na 2 CO 3 (соде) или по тетраборату натрия Na 2 B 4 O 7 . 10Н 2 О (буре). Бура при растворении взаимодействует с водой:
В 4 О 7 2– +3Н 2 О=2Н 3 ВО 3 + 2ВО 2 –
Образовавшийся метаборат - довольно сильное основание. Его титруют кислотой:
ВО 2 – + Н 3 О + = Н 3 ВО 3 .
Очевидно, что молярная масса эквивалента буры равна М (½Na 2 B 4 O 7 . 10Н 2 О) = 190.71 г/моль. Высокая молярная масса эквивалента – преимущество буры как первичного стандарта. Растворы щелочей стандартизуют по гидрофталату калия. Молекулагидрофталатасодержит подвижный протон и обладает свойствами слабой кислоты:
В качестве стандартов нередко используют бензойную кислоту С 6 Н 5 СООН, щавелевую кислоту H 2 C 2 O 4 . 2H 2 O и другие слабые органические кислоты (твердые, чистые устойчивые вещества). Стандартные 0,1000 М растворы кислот и оснований в лабораториях обычно готовят из фиксаналов. Приготовленный раствор кислоты можно использовать для стандартизации раствора щелочи, и наоборот. Стандартизованные растворы кислот устойчивы и могут храниться без изменения сколь угодно долго. Растворы щелочей менее устойчивы, рекомендуется хранить их в парафинированной или фторопластовой посуде, чтобы не допустить взаимодействия со стеклом. Необходимо учитывать, что растворы щелочей поглощают СО 2 из воздуха, при хранении их защищают с помощью трубки, заполненной негашеной или натронной известью.

Рис. 2. Кривые нейтрализации сильной кислоты.
1 - 0,1 М, 2 - 0,01 М, 3 – 0,001 М.
Для обнаружения к.т.т. с цветным индикатором необходимо, чтобы высота скачкабыла больше, чем ширина интервала перехода индикатора. Последняя обычно составляет около двух единиц рН.
Высота скачка на кривой нейтрализации слабых кислот зависит отсилы кислоты(величины ее кислотной константы, или рK a ). А именно, чем слабее кислота (чем больше величина рК а), тем меньше при прочих равных условиях должны быть высота скачка.разной силы
1 -соляная кислота,2 – уксусная кислота (рК а = 4,8),3 – синильная кислота (pK a = 9,2).
Высота скачка должна быть большеширины зоны перехода индикатора, которая, как правило, составляет 2 единицы рН. Поэтому,как и в случае сильных электролитов, критерий возможностититрования слабого протолита с 1 %-ной ошибкой можно вывести из условия ∆p Н ±1% ≥ 2. Для водного раствора слабой кислоты получаем искомый критерий в следующей форме:
рК a + рС ≤ 8
Приp С = 2критическое значение рК а равно 6. Иными словами, если кислота очень слабая, и ее рК а больше 6, то точно оттитровать ее с цветными индикаторами нельзя.
Титрование смесей протолитов и многопротонных протолитов. В смешанных растворахсильные кислоты подавляют протолиз более слабых. То же наблюдается в растворах, содержащих смесь оснований разной силы.При добавлении к такой смеси титранта прежде всего оттитровывается более сильный протолит, а уже затем с титрантом реагирует более слабый. Однако число скачков, наблюдаемых на кривой титрования смеси, зависитне только от числа присутствующих протолитов, но и от абсолютных значений соответствующих констант кислотности (основности), а также от их соотношения. Константы кислотности (или основности) компонентов смеси должны различаться более чем в 10 4 ,раз, только в этом случае на кривой титрования будутраздельно наблюдатьсяотчетливо выраженные скачки титрования, а относительная ошибка определения каждого компонентане превысит 1 %. Критерием возможности раздельного титрования протолитов является так называемое «правило четырех единиц»:
![]() (6)
(6)
Многопротонные протолиты реагируют с титрантамиступенчато, сначала по первой ступени, затем по второй и т.д., если соответствующие константы кислотности различаются в соответствии с условием (6).При расчете кривых нейтрализации многопротонные протолиты можно рассматривать каксмесиразных электролитов.
В качестве примера проанализируем возможность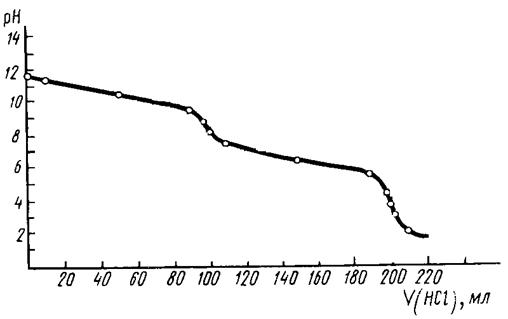
Рис.5. Кривая титрования смеси карбонат- и гидрокарбонат-ионов раствором HCl .
Указаны значения рН, при которых наблюдаются переходы окраски индикаторов.
При титровании смеси двух сильных кислот, смеси двух одинаково слабых кислот или смеси двух оснований с близкими рК b двух раздельных скачков на кривой титрования нет. Однако определить концентрацию компонентов таких смесей по отдельности все же вполне возможно. Эти задачи успешно решают, используя дифференцирующие неводные растворители.
Кислотно-основные индикаторы и их выбор
Для обнаружения к.т.т. в методе нейтрализации традиционно используют кислотно-основные индикаторы – синтетические органические красители, являющиеся слабыми кислотами или основаниями и меняющие видимую окраску в зависимости от рН раствора.Примеры некоторых (наиболее часто применяемых в лабораториях) кислотно-основных индикаторов приведены в таблице 3. Строение и свойства индикаторов приведены в справочниках. Важнейшими характеристиками каждого кислотно-основного индикатора являются интервал перехода и показатель титрования (pT ). Интервал перехода – это зона между двумя значениями рН, соответствующими границам зоны, внутри которой наблюдается смешанная окраска индикатора. Так водный раствор метилового оранжевого наблюдатель охарактеризует как чисто желтый – при рН< 3,1 и как чисто красный при рН > 4,4, а между этими граничными значениями наблюдается смешанная, розово-оранжевая окраска разных оттенков. Ширина интервала перехода обычно составляет 2 единицы рН. Экспериментально определенные интервалы перехода индикаторов в некоторых случаях меньше или больше двух единиц рН. Это, в частности, объясняется различной чувствительностью глаза к разным участкам видимой области спектра. Для одноцветных индикаторов ширина интервала зависит и от концентрациииндикатора.
Таблица 3
Важнейшие кислотно-основные индикаторы
|
Индикатор |
Интервалперехода ΔрН Ind |
рК a (HInd ) |
Изменение окраски |
|
|
Метиловый оранжевый |
Красная - желтая |
|||
|
Бромкрезоловый зеленый |
Желтая - синяя |
|||
|
Метиловый красный |
Красная - желтая |
|||
|
Бромкрезоловый пурпурный |
Желтая - фиолетовая |
|||
|
Бромтимоловый синий |
Желтая - синяя |
|||
|
Феноловый красный |
Желтая - красная |
|||
|
Тимоловый синий |
||||
|
Фенолфталеин |
Бесцветная - красная |
Зная характеристики разных индикаторов, можно теоретически обоснованно подбирать их,чтобы получить правильные результаты анализа.Придерживаются следующего правила: интервал перехода индикатора должен лежать в области скачка на кривой титрования .
При выборе индикаторов для титрования слабых протолитов необходимо учитывать, что т.экв. и скачок титрования смещены в слабощелочную среду при титровании кислоты и в слабокислую среду – при титровании основания. Следовательно, для титрования слабых кислот подходят индикаторы, меняющие свою окраску в слабощелочной среде (например, фенолфталеин), а для титрования слабого основания – индикаторы, меняющие окраску в слабокислой среде (например, метиловый оранжевый
Существует ещё одна характеристика каждого кислотно-основного индикатора –это показатель титрования (рТ). Так называют значение рН, при котором наблюдатель наиболее отчетливо замечает изменение окраски индикатора и именно в этот момент считает титрование законченным. Очевидно, рТ = рН К.Т.Т. .Выбирая подходящий индикатор, надо стремиться к тому, чтобы величина рТ была бы как можно ближе ктеоретически рассчитанной величине рН Т.ЭКВ.. Обычно значение рТблизко к середине интервала перехода. Но рT – плохо воспроизводимая величина. Разные люди, проводящие одно и то же титрование с одним и тем же индикатором, получат существенно различные значения pT .К тому же величина рТ зависит от порядка титрования, то есть от направления изменения окраски.При титровании кислот и оснований с одним и тем же индикатором значения рТбудут несколько различаться. Для одноцветных индикаторов (фенолфталеин и т.п.) величина рТ зависит и от концентрации индикатора.
Заполненной титрантом до нулевой отметки. Титровать, начиная от других отметок, не рекомендуется, так как шкала бюретки может быть неравномерной. Заполнение бюреток рабочим раствором производят через воронку или с помощью специальных приспособлений, если бюретка полуавтоматическая. Конечную точку титрования (точку эквивалентности) определяют индикаторами или физико-химическими методами (по электропроводности, светопропусканию, потенциалу индикаторного электрода и т. д.). По количеству пошедшего на титрование рабочего раствора рассчитывают результаты анализа.
Виды титриметрического анализа
Титриметрический анализ может быть основан на различных типах химических реакций:
- кислотно-основное титрование - реакции нейтрализации ;
- окислительно-восстановительное титрование (перманганатометрия, иодометрия , хроматометрия) - окислительно-восстановительные реакции ;
- осадительное титрование (аргентометрия) - реакции, протекающие с образованием малорастворимого соединения, при этом изменяются концентрации осаждаемых ионов в растворе;
- комплексонометрическое титрование - реакции, основанные на образовании прочных комплексных соединений ионов металлов с комплексоном (обычно ЭДТА), при этом изменяются концентрации ионов металлов в титруемом растворе.
Типы титрования
Различают прямое, обратное титрование и титрование заместителя.
- При прямом титровании к раствору определяемого вещества (аликвоте или навеске, титруемому веществу) добавляют небольшими порциями раствор титранта (рабочий раствор).
- При обратном титровании к раствору определяемого вещества добавляют сначала заведомый избыток специального реагента и затем титруют его остаток, не вступивший в реакцию.
- При заместительном титровании к раствору определяемого вещества добавляют сначала заведомый избыток специального реагента и затем титруют один из продуктов реакции между анализируемым веществом и добавленным реагентом.
См. также
Ссылки
Wikimedia Foundation . 2010 .